
本文来自微信公众号:同写意(ID:tongxieyi),作者:万里(启德医药高级副总裁和国际注册事务部负责人),审稿:应嘉、夏毅、洪筱玲、黄桂林、杜新,原文标题:《FDA的这堂公开课到底讲了什么?——信达/礼来ODAC会议的启示》,头图来自:IC photo
美国时间2月10号,备受瞩目的美国食物药品监督管理局(FDA)为信达/礼来PD-1抗体信迪利单抗(sintilimab)与化疗联用作为非小细胞肺癌一线疗法的新药上市申请(BLA)的肿瘤药物专家委员会议(ODAC会议)如期召开。
经过5个多小时的讨论并投票表决,肿瘤药物专家委员会最终以14票赞成对1票反对的压倒性投票支持FDA的提议,要求信迪利单抗在获批前需要补做新的临床试验。笔者虽然对于这次ODAC投票结果感到遗憾,但是认为信达作为中国生物制药公司中的领头羊勇于出海,成为第一个吃螃蟹与FDA开ODAC会议的中国创新药企业,这种挑战自我的精神还是值得肯定的。
FDA通过这次的ODAC会议向所有人尤其是中国的生物制药公司明确了对于来自中国的单一国家临床数据到美国申请新药上市的准入要求,从ODAC会议的结果也可以看出来中国的生物制药公司在对于国际监管机构的注册要求和政策变化的理解,全球注册策略的制定,临床设计和执行等方面与FDA的要求还是有不小的差距。
对于信达生物而言,这次的ODAC会议结果无疑让信迪利单抗美国上市和中国PD-1产品出海暂时受挫,但对于国内众多致力于创新药研发的公司来说,却是由FDA免费上了一次宝贵的公开课。而对每个有创新药出海的国际化雄心,并希望早日构筑国际竞争优势的中国生物医药公司来说,熟悉FDA等国际监管机构监管规则和要求,懂得如何与FDA沟通,了解如何设计和执行MRCT临床试验,更是迫切需要的必修课。
那么FDA在ODAC会议这堂公开课上到底讲了什么?而准备出海的中国药企又可以从这堂公开课上学到什么?笔者根据多年的国际注册经验,对于这次ODAC的会议内容和FDA的审评要求进行深入解读,希望能为准备出海的中国药企的提供一些有益的帮助。

一、新药研发需要从早制定全球注册策略,并且与时俱进、贯穿始终
一个新药的开发,平均需要超过10亿美金规模的资金投入以及10到15年的时间投入,才能在新药全球上市后得到商业回报。新药开发的临床周期如此长,花费如此高,所涉及的因素又如此多,新药研发如何确保临床开发方向的最优化?如何做出科学合理的判断并决定继续还是终止临床试验?怎么确保临床数据满足监管机构的上市要求?
一般来说,为了提高新药研发的成功率,任何一个新药的研发一旦进入临床阶段,就需要制定一个非常全面合理的临床开发计划(CDP,Clinical Development Plan)和相应的全球注册策略(GRS,Global Regulatory Strategy)。
临床开发计划是一个新药临床开发的整体计划,而全球注册策略则是支持临床开发,并在全球成功获批上市,并达成目标产品概况(TPP,target product profile)的蓝图。
全球注册策略一般按照以终为始的方法,根据产品的最终临床需求(比如产品说明书product label)和监管机构的上市要求来反推需要的临床数据,根据不同地区监管差异和特点来确定每个地区的最佳申报途径,由一个包括临床、注册、 CMC、非临床、市场、和医学事务的跨领域团队(cross-functional team)共同完成。
欧美的药企传统上会先考虑欧美日的上市要求来制定这在这三个主要市场上市的注册策略,随着中国成为世界第二大药品市场并在2017年加入ICH,过去几年中国监管政策与国际逐渐接轨,新药临床申请(IND)审评时间也缩短到了大概三个月,使得越来越多的欧美公司在一开始制定国际注册策略时,就会考虑在中国做桥接试验或者MRCT临床试验以加快在中国的上市速度。
尤其是在2017年后,ICH推出了E17文件来指导如何使用MRCTs的平行全球注册策略来替代之前的E5文件中用桥接试验来外推其他地区临床试验结果的策略。国际多中心试验可以更有效的提高临床开发速度,避免临床资源的浪费,明显优于单一国家临床加上不同地区桥接试验的做法。
而对于把目标定位在国际新药研发和海外产品上市的中国药企,从一开始制定注册策略就应该考虑中国以外地区,尤其世界第一大药品市场——美国的上市要求,通过MRCT来加快在中国以及海外的上市速度。
值得一提的是,新药产品一旦获得了美国FDA的上市批准,也意味着得到了世界上最高水平的监管机构的认证,就可以免去新的临床试验而通过药品证书CPP(Certificate of Pharmaceutical Product)申报的方式在很多接受FDA药品证书的其他国家和地区(比如亚洲,拉丁美洲,南美洲,中东和非洲)获批上市,也就是说FDA批准不仅是打开美国市场的钥匙,也是打开世界市场的钥匙。
在ODAC会议上,信达/礼来提到ORIENT-11一开始只是计划在中国上市批准,所以临床设计也是按照中国的注册要求来做的,但是在2020年初获得ORIENT-11临床试验的中期分析数据后,同时看到2019年FDA肿瘤学卓越中心主任Richard Pazdur博士在AACR年会上表示完全来自中国数据的药物只要质量足够高就可以到美国申请上市,才决定向FDA提交了ORIENT-11临床试验的结果并申请上市批准。
从注册的角度来看,信迪利单抗的开发本来只是考虑了中国注册要求可以理解,但是缺乏一个完整全面的全球注册策略难免给产品的出海之路带来很多一开始预料不到的困难,也造成了ORIENT-11的临床设计和结果与FDA的注册要求存在许多差距。
除了熟悉FDA的注册要求,中国药企也需要密切关注每个国家尤其是美国监管政策的变化,肿瘤药的开发竞争激烈,临床进展更是日新月异,所以任何一个新药的临床开发计划和全球注册策略都需要与时俱进,随时了解竞品开发和监管政策的改变,从而做出相应的变化。
Richard Pazdur博士也在ODAC会议的最后决议之前特意发言解释了说他在AACR年会上发言之后改变想法的原因,提到了很多因素都有了改变,所以新药的开发策略应该以与FDA的充分沟通为准,而不应该仅凭他在某个公开场合的讲话。
诚如Richard Pazdur博士所说,美国的监管政策最近几年确实有很多新的变化,第一是FDA对于PD-1产品的批准正在收紧。FDA过去7年已经批准了7个PD-1/PD-L1的抗体药品,超过85种的适应症,其中很多是以替代终点批准的加速上市。所以FDA在去年四月特地召开ODAC会议重新评估是否继续批准未在验证性试验中显示出临床疗效的三大PD-(L)1的六个加速批准适应症,并在去年共撤回了7个肿瘤药的加速批准适应症,还建议Agenus撤回其PD-1单抗balstilimab用于化疗后疾病进展的复发或转移性宫颈癌患者的上市申请。
今年一月赛诺菲(Sanofi)和再生元(Regeneron)也由于未能与FDA就上市后研究达成一致而撤回了PD-1抗体Libtayo二线治疗晚期宫颈癌患者的上市申请(sBLA)。同时在非肿瘤领域,2021年给予Biogen公司治疗阿尔茨海默症的药物Aduhelm上市批准就引起了很大的争议,也让FDA受到了公众很大的压力,觉得其对于新药的上市批准尺度太松。
第二是FDA确实在大力倡导Project Equity,并在2020年推出了提高临床试验人群多样性的指导原则,希望把黑人患者在内的美国少数族裔包括到临床研究中,FDA在过去两年中几乎所有新的NDA/BLA批准中都要求上市后临床研究PMC中纳入黑人患者。
考虑到礼来作为一家老牌的美国药企,从2015年就与信达合作,直到2020年签订协议买下信迪利单抗的海外权益,所以理论上礼来应该凭借其对于美国监管政策的熟悉给以信达指导,对于这个结果礼来显然难辞其咎。
不出意料的是FDA在ODAC会议上把矛头主要对准了信达生物在美国的合作伙伴礼来,Richard Pazdur博士在会议上质问礼来为什么在承诺增加试验多样性的同时,用这样一个完全来自中国的单一国家临床数据来提交上市申请。甚至在讨论中提到双方沟通时间的陈述中出现了一些充满火药味的语句,礼来声称在提交BLA之前双方沟通过三次。
FDA肿瘤二科主任Harpreet Singh博士则表示FDA一直到2020年4月的pre-IND会议中才知道信达做了ORIENT-11临床试验并准备申报BLA,并在与信达的沟通中提出了非常多的担忧,而礼来意外的公布了一页2020年8月与FDA的Pre-BLA的会议纪要,也让FDA负责人非常恼火,甚至提出要向公众公开所有的沟通文件,礼来只有在会上进行道歉,表示无意曲解与FDA的沟通和误导ODAC委员会。
一般来说,选择在ODAC会议上与FDA这样针锋相对的激烈讨论需要非常慎重,因为FDA大多是在产品的获批存在重大争议或者有很大不确定性的时候才会召开ODAC会议。有经验的药企还是需要从临床开始阶段就与FDA保持良好的沟通和信誉“credibility”,让FDA对你的公司,产品和数据都有信心,在有争议的时候尽量通过良好沟通来解决,而不要选择ODAC会议这样的方式。
由于ORIENT-11试验本来就是按照中国上市的要求进行设计的,没有考虑到美国上市的要求,所以出现很多的硬伤也并不奇怪。如果只是一两个问题,那么还是有可能与FDA通过协商PMC/PMR来解决,因为FDA的审评原则是看证据链总体性“totality of evidence”,可是太多的硬伤就很难通过沟通来弥补。
相信如果信达/礼来与FDA尽早进行沟通,获得FDA对于注册研究临床设计的反馈及时调整临床方案修补硬伤,而不是等到药物提出上市申请的最后一刻才进行,那么这次ODAC会议的结局也许会不一样。
而在与监管机构沟通的时候还需要考虑到中美两国监管机构的风格的不同,CDE像一个教练,会直接指导企业应该在研发中做哪些工作,而FDA更像一个裁判,不会直接指导企业怎么做,而是希望企业提出自己的开发想法后给予具体的判断意见。所以企业在设计临床试验尤其是申报上市关键临床的时候就需要充分与FDA沟通,获得FDA的认可后再开始临床试验,这样可以帮助企业少走很多不必要的弯路。
而在与FDA在新药上市申请中的沟通更是一项复杂而繁琐的系统工程,需要通过pre-NDA/BLA meeting,orientation meeting,mid-cycle communication, late-cycle meeting等一系列审评会议与FDA保持良好沟通,及时就FDA的问题和发补完成答复,避免新药审批的延误,确保早日获批上市。
总而言之,新药研发需要从早制定全球注册策略,并且与时俱进贯穿始终,同时与FDA建立起畅通的沟通交流,让FDA对所有的新药研发和临床试验进展保持知情,这将大大提高新药临床和上市获批的成功率。
二、临床试验设计是一项需要跨部门合作的工作
众所周知,临床试验的方案设计是一项需要跨部门合作的工作,一个高质量的临床试验设计离不开医学,临床运营,临床药理,数据管理、生物统计,临床安全,和注册等各个部门的紧密配合和综合考虑。
ORIENT-11试验的多个硬伤其实是在试验设计就有的问题,当初临床设计可能只是简单地照搬了Keynote的试验设计,满足了当时注册策略中的最低要求,没有让有经验的统计和注册来进行把关。
FDA在ODAC会议上没有质疑ORIENT-11试验的有效性和安全性,但是提出导致其数据无法推广到美国人群的多个缺陷,主要包括:主要终点采用了PFS而不是OS,使用了化疗而不是已经上市的免疫疗法做对照组,所有入组病人都是中国人而没有欧美人种的数据,知情同意书没有按照美国要求及时更新,以及可能存在缺乏数据完整性的问题。
如果信达在这个临床试验启动或者进行中与FDA沟通,FDA会要求把对照组改成已经上市的Keytruda加上化疗,因为在入组第一个患者前这个组合在美国已经批准用于一线肺癌,也会建议用OS而不是PFS作为终点。信达答复中提到ORIENT-11本来只是计划在中国上市,所以整个设计当时只是考虑满足中国的注册要求,而中国CDE的指导原则《晚期非小细胞肺癌临床试验终点技术指导原则》中明确提出可以接受单独PFS作为晚期NSCLC注册研究的主要终点。
迄今为止,FDA对转移性非小细胞肺癌一线免疫治疗(IO,Immuno-Oncology therapy)产品的所有上市完全批准(full approval)都是基于总体生存OS的统计学上的改善。免疫治疗相对于靶向治疗和化疗的优势主要体现在OS生存期的延长,所以TKI药物的批准是可以用PFS作为终点的,但是所有PD-1药物的完全批准都要求用OS作为终点。
Keytruda的Keynote-189三期临床一开始也是用PFS做主要终点,但是后来加上了OS作为主要终点,在试验成功后获得了非小细胞肺癌一线治疗的完全批准上市。
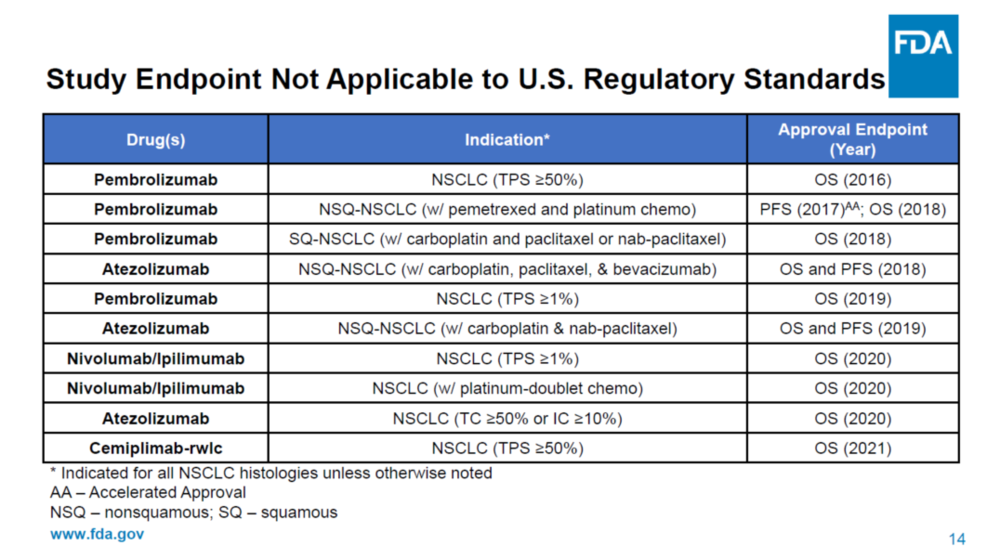
ODAC会议上也讨论到在没有预先设定严格的OS分析的情况下,怎么评价ORIENT-11的OS数据?信达/礼来解释说,虽然ORIENT-11没有事先定义严格的OS假设检验,但是在PFS中期分析之前已经有了OS观察的计划,然后也有通过方案修订将最终时间定在最后一个患者被随机入组后大约两年,并在会议上展示ORIENT-11的post-hoc OS分析结果是有效而稳健的。
但是信达/礼来的这个解释显然非常无力,如果没有预先设定的OS分析,包括分配alpha值、多重性校正以及定义OS目标事件数等,而只是做post-hoc分析得出的OS数据只是描述性的,缺乏统计的严谨性。就像没有在开枪之前就画好靶子,无论之后打的有多准都无法说明射击的准确性。
FDA也明确回应这些分析只能支持假设生成hypothesis generating,不能用于假设验证hypothesis testing。如果对于FDA的审评特点有所了解,就会知道FDA对于关键临床试验的设计和试验执行过程中数据完整性和统计严谨性的重视。
FDA有一个非常大的统计部门负责统计审评,在关键临床试验之前会对试验设计提出指导意见,在新药申请时要求申请人提供所有的临床试验数据,由FDA的统计师来独立计算,确认所有的结果。
笔者认为如果信达在试验设计初期把OS作为一个关键次要终点(key secondary endpoint),采用gatekeeping守门法的方式,在PFS分析成功之后对OS做分析,并不会消耗alpha值影响试验成功,也不增加整个试验的复杂度,反而可以额外获得严格意义上的OS结果。
值得一提的是,新药上市虽然是BLA的主要目的,但是更重要的是新药最终能够成功launch product让病人获益并获得商业上的成功,与FDA就产品说明书Product Label的讨论是重中之重,因为美国的医药销售和医生开药的时候最重视的就是Label上的有效性和安全性说明。
Label上的每一条声明都是需要用临床数据来支持,并在临床试验设计的时候就考虑到这些需求,所以一般有经验的制药公司会在设计关键2/3期临床试验的时候,即使只是用OS作为次要终点(secondary endpoint),也会给出统计假设并事先分配alpha值,因为这样得到的OS的数据就可以放到Label上,在市场上已经有很多竞品的情况下更有机会被医生青睐,从而促进病人使用和产品销售。
三、临床执行的细节决定试验的质量
除了与FDA沟通和临床设计等问题,FDA对于 ORIENT-11试验在试验人群选择,伦理的合规性,和临床数据的完整性也提出了质疑。
FDA称这个ORIENT-11用于支持审批的三期临床试验完全在中国进行,而不是全球多中心临床试验,因此并未反映美国肺癌患者的种族和民族多样性,不再符合Project Equity的精神,接受此类研究与行业范围内对临床试验公平性、多样性的承诺存在冲突。
但是这并不表示,FDA完全不接受海外临床数据包括中国数据,其实FDA对于海外数据的接受政策已经在21 CFR 312.120and314.106中给出了非常详细的规定,需要达到三个要求:
1. 国外数据适用对美国人口和医疗实践;
2. 研究已经由具有公认能力的临床研究人员进行;
3. FDA可以通过现场检查或其他适当方法验证临床数据的可靠性。
之前也不乏用海外数据获批上市的产品,比如2019年百济神州的泽布替尼Brukinsa基于在中国的关键性二期BGB-3111-206试验数据获得FDA加速批准上市用于复发难治性套细胞淋巴瘤的治疗,2021年日本第一三共制药(Daiichi Sankyo)靶向HER2的抗体药物偶联物(ADC)药物Enhertu基于在日本和韩国的关键二期DESTINY-Gastric 01试验的结果获得FDA批准用于治疗已接受过曲妥珠单抗治疗的局部晚期或转移性HER2阳性胃癌或胃食管交界腺癌病人。
当然这两个适应症在美国都是罕见病,而这两个产品也都显示了非常显著的临床疗效,满足了FDA对于海外数据的接受要求,所以FDA给予相应的监管灵活性批准上市。
FDA对于信迪利单抗试验的对照组选择也表现出极大不满,因为这涉及到伦理审查问题。FDA在讨论中提到如果信达提前与他们沟通的话,FDA肯定会要求把对照组改成K药+化疗做非劣效研究来满足要求,因为在入组第一个患者前这个组合在美国已经批准用于一线肺癌。
Harpreet Singh博士对礼来发出质疑:“礼来对剥夺患者延长总体生存期的治疗的化疗组感到满意吗?礼来进行了多少次试验,剥夺了患者获得已知生存优势的治疗方法?”Richard Pazdur博士也表示:“我对这个问题感到非常不舒服,因为已知疗法可以改善一年以上的平均生存率,而患者却没有得到它。我希望委员会成员就这部分进行一些讨论。”
笔者认为FDA在这个问题上有点苛求,考虑到ORIENT-11临床试验启动时K药并未在中国获批上市,采用化疗作为对照组也可以理解。
FDA还强调了ORIENT-11试验在知情同意程序方面的疏忽,尤其是在2019年3月Keytruda获得中国药监局批准上市联合化疗用于一线非小细胞肺癌之后,知情同意书在试验期间没有按照要求更新,让病人获知有新的免疫治疗产品获批上市。知情同意程序没有让病人获得充分地知情权,也不符合美国临床实践。ODAC会议上专家也认为,ORIENT-11试验未能达到FDA的知情同意标准,因为它没有明确列出经批准的疗法或参与替代研究的治疗方法。
美国国家癌症研究所临床主任Ravi Madan教授表示:“虽然数据完整性在临床研究中至关重要,但伦理方面的完整性更为重要。”信达生物和礼来也在会议上表示已经认识到在知情同意程序上的不足,并已经更新了内部的知情同意流程,确保在病人入组时提供完整的信息,让病人明确了解当时已有的治疗方法,可以自己判断是否同意入组临床研究。
FDA在ODAC会议上指出在中国研究者参加国际多中心的临床试验经验有限,还直接引用中国食品药品监督管理局2016年报告,称80%的中国临床研究存在造假或不合格。事实上自2015年7.22临床数据核查后,临床试验的真实性、规范性和完整性一直是中国新药研发重点监管对象,中国新药临床研究已与国际接轨,数据不真实已几乎不存在。FDA对ORIENT-11试验的48个临床中心中的两个中心进行GCP核查也证实了没有发现任何数据完整性的问题。
笔者认为,在这个问题上,FDA援引6年前的报告的做法显然是对于中国临床有些刻板印象之嫌,信达的回应非常有力,澄清了这些年来中国临床研究在试验质量、数据完整性和监管都有所提高的事实,让大家了解到中国临床研究现在是能达到国际标准的。
但是FDA对于ORIENT-11试验在人群选择,伦理的合规,和临床数据的真实性的质疑提醒了中国企业将来在做临床研究时,一定要更加注重临床执行的细节,一开始就按照国际标准做好临床设计和执行,以保障临床试验的质量和GCP合规性。
四、中国PD-1产品美国上市还有机会吗?
FDA非常罕见地在这次的ODAC会议之前在新英格兰医学杂志(NEJM)和柳叶刀(Lancet)等不同的专业期刊和媒体上发表了几篇文章,指出美国和世界范围内PD-1/PD-L1抗体的过度开发的状况,虽然FDA已经批准了7个PD-1/PD-L1抗体,但是FDA仍然收到了大约有25个来自中国公司PD-1/PD-L1抗体的新药上市申请,而且这些申请几乎都是只有中国数据。
可以说FDA在这次ODAC会议之前已经对于信迪利单抗的上市申请定了基调,但是FDA显然还是希望通过这次的ODAC会议向所有人尤其是中国的生物制药公司,明确对于来自中国的临床数据到美国申请新药上市的准入要求。
虽然说ODAC会议投票意见不具有对FDA决策的约束力,但是考虑到FDA在这次ODAC会议之前和会议上的表态,基本上已经可以推断,等3月22号PDUFA日到期时,FDA大概率会给出一封拒绝批准上市的完整回应函(CRL,complete response letter)。
平心而论,FDA对于信迪利单抗的评估还算公允,ORIENT-11试验的主要终点,对照组选择,研究人群,以及知情同意程序等方面确实与FDA的要求存在差距,而且没有在临床试验前就临床设计与FDA沟通达成一致。
虽然信迪利单抗很有可能在种族之间没有差别,疗效理论上可以转化到美国人群,但是没有直接的临床数据证明这一点。最关键是FDA在转移性非小细胞肺癌一线治疗这个适应症上已经批准了10个PD-1/PD-L1单药或联合用药产品,信迪利单抗不能解决未满足的临床需求。应该说FDA的这个审评原则与中国CDE倡导的以临床价值为导向的精神是完全一致的,都是鼓励更新或更好、能够解决未满足临床需求的创新药研发。
所以这个ODAC会议结果并不代表FDA完全关上了新药产品用中国临床数据在美国上市的这个大门,FDA在会议上明确表示只要满足FDA对于海外数据的接受要求,针对未满足的临床需求(unmet medical needs),罕见病(MRCT难以开展)、全创新药等3种情况还是会给予相应的监管灵活性,比如鼻咽癌(NPC,Nasopharyngeal Carcinoma)和儿童癌症,在美国属于罕见病,也没有已经批准的有效治疗,那么还是存在用中国的临床数据获得美国批准上市的可能性。
在ODAC委员会投票阐明了立场之后,现在在信达面前好像只有一条可行的路径,那就是与已经上市的PD-1产品(比如K药)用OS做终点做一个头对头的非劣效MRCT试验来证明信迪利单抗在美国病人中的疗效不劣于K药。但是这样的开发路径不仅投资高,时间长,在美国招募病人会有很大挑战,而且还具有非常大的不确定性,所以感觉信达/礼来可能不会采取这样的路径。
其实信达/礼来可能是基于商业上的考虑,选择了非小细胞肺癌一线治疗这样一个在美国已经有很多PD-1产品获批上市、门槛非常高的适应症申报BLA,但是从ODAC会议的结果来看,这种选择还是值得商榷的。
那么是否有其他的路径?我觉得在ODAC会议上,FDA其实反复强调对于未满足的临床需求还是会给予相应的监管灵活性,所以更为可行的一条路径应该是像君实和康方申请鼻咽癌的适应症一样,找到一个罕见病、具有未满足的临床需求、而且没有已经批准的PD-1产品的适应症来获得上市批准,然后通过降低价格和扩大标签使用来获得更多的市场份额。
而在信达的信迪利单抗之后,还有3款国产PD-1均已于去年提交美国BLA申请,今年等着排队上市了,分别是君实生物的特瑞普利单抗,康方生物的派安普利单抗和百济神州替雷利珠单抗。替雷利珠单抗拥有全球多中心MRCT试验数据,而特瑞普利单抗和派安普利单抗则是瞄准了鼻咽癌的适应症,理论上能够满足FDA的准入要求或者获得监管灵活性,相信2022年肯定会听到中国的PD-1产品在美国获批上市的好消息。

五、结束语
FDA通过这次ODAC会议给中国生物制药企业上的这堂公开课可能最大的作用就是明确给出了中国新药到美国上市的准入要求,让许多正在用中国数据申报BLA的中国公司的美国梦可能随之破灭,但是也给认真做药具备实力出海的新药公司提供了一个明确的路径:
从临床1期开始就做好国际注册策略和临床开发计划,构建一个包括医学,临床运营,临床药理,数据管理、生物统计、临床安全和药物警戒,和注册在内的跨部门团队,进一步熟悉国际监管要求,通过与国际监管机构的沟通和合作,开展MRCT国际多中心临床试验,开发出真正能够解决中国和欧美未被满足临床需求的新药。
FDA在ODAC会议前也提出希望把中国纳入FDA发起并主导的多国同步平行审评计划Project Orbis,这样可以促进MRCT的发展,加快创新肿瘤药在全球主要市场同步上市的速度,当然这对于中国的本土创新企业提出了更高的要求,毕竟在欧美药企的研发经验和水平还是要高于中国药企的情况下,全球加速上市也意味着对于中国药企的更大挑战。
但是国际化是中国创新药发展的必然路径,预计本次事件将进一步刺激中国创新药企加快出海参与全球竞争的步伐,未雨绸缪早日构筑国际竞争优势,提升国产创新药研发实力。
信达/礼来这次与FDA的ODAC会议长期来看肯定会对中国生物制药公司的国际化和成功出海起到促进的作用,成为中国创新药发展历史上的一个重要里程碑。整个过程可以用北京2022年冬奥会上,羽生结弦在男子单人自由滑中未能成功挑战阿克塞尔四周跳摔倒之后,央视解说员陈滢为他赋诗中写的这段话来形容:“守一座守不住的城,打一场打不赢的仗,天意终究难参,假若登顶成憾,与君共添青史几篆,成败也当笑看。”
特别感谢:罗氏基因泰克公司全球注册高级总监应嘉博士,启德医药生物统计副总裁夏毅博士,安博生物法规事务副总裁洪筱玲博士,Relay Therapeutics法规事务副总裁黄桂林,埃格林医药CEO杜新博士在百忙之中对本文进行了认真审阅,给予了宝贵意见。
参考文献
[1] Beaver, J. A., & Pazdur, R. "Dangling" Accelerated Approvals in Oncology, N Eng J Med. 2021 May 6.
[2] Beaver, J. A., & Pazdur, R. The Wild West of Checkpoint Inhibitor Development. N Engl J Med. 2021 Dec 15.
[3] Singh, H., & Pazdur, R. Importing oncology trials from China: a bridge over troubled waters? Lancet Oncol. 2022 Feb 4
[4] Steven Lemery , & Richard Pazdur, Approvals in 2021: dangling Accelerated Approvals, drug dosing, new approvals and beyond, Nat Rev Clin Oncol. 2022 Feb 8;1-2.
[5] Adam Feuerstein, ‘I have a right to change my mind’: A top FDA regulator is unapologetic over his about-face on Chinese cancer drugs, STAT Feb. 8, 2022
本文来自微信公众号:同写意(ID:tongxieyi),作者:万里 博士(启德医药高级副总裁和国际注册事务部负责人、美中药协注册社群负责人)