
本文来自微信公众号:学术经纬 (ID:Global_Academia),作者:药明康德内容团队,头图来源: 视觉中国,原文标题:《新的里程碑!〈自然〉重磅专题:首个人类泛基因组草图公布》
今日,人类遗传学研究迎来了又一里程碑:首个人类泛基因组参考(human pangenome reference)草图发布!

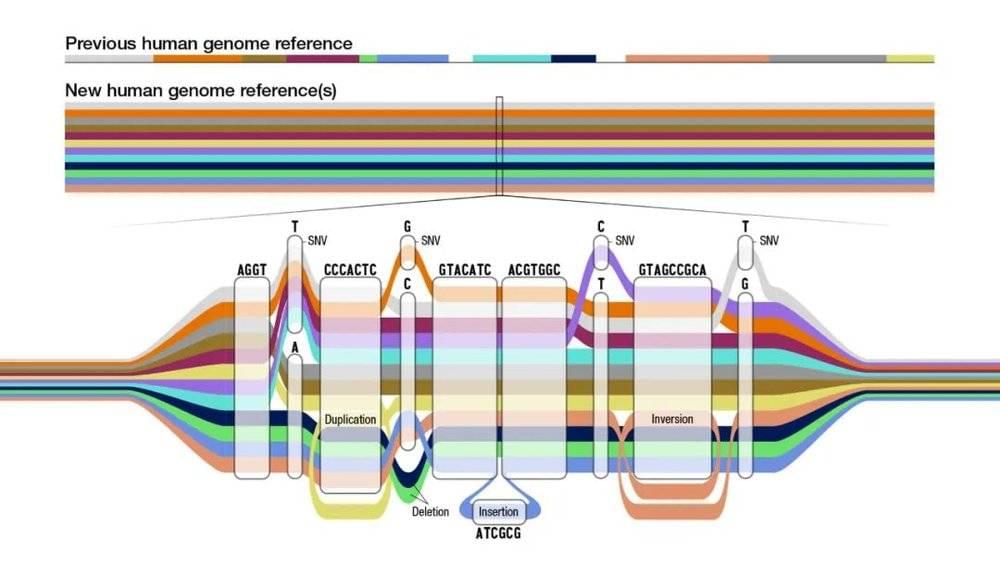
这份泛基因组参考草图的问世,意味着我们将更全面、更准确地了解全世界人类的基因组多样性。最新图谱结合了来自不同祖源的47个个体的遗传信息,比现有的单一参考基因组序列(GRCh38)新增1.19亿个碱基字母。该图谱为人类基因组提供了更完整的图像,也更能代表人类这个物种的遗传多样性,应用这一参考图谱能极大提高对人类基因组中变异体的检测。
顶尖学术期刊《自然》同时发表了3篇论文,另有第4篇论文发表于《自然-生物技术》。在这一专题中,人类泛基因组参考联盟(Human Pangenome Reference Consortium)提出了首个人类泛基因组参考草图的构建和使用方法,并报告了使用该图谱获得的两项新发现。

从1到全世界
21世纪之初,人类基因组计划(Human Genome Project)发布了第一版人类参考基因组草图,标志着解读人类生命蓝图的突破性里程碑。然而由于当时的测序技术所限,这版草图中留有许多空白。
最初的参考序列在此后的二十年里不断更新,包括修复错误和填补空白。2003年,人类基因组计划产生了占人类基因组90%以上的序列。然而直到2019年,人类基因组测序结果中仍有数百万个碱基位置上是空白的。
就在去年,填补人类基因组空白的工作取得重大突破。由于长读长和超长读长测序技术的发展,研究者读取生物样本的DNA时可以一次性解码几千甚至百万个碱基对,然后通过专门的算法将这些DNA长片段组装成更完整的基因组序列。“端粒到端粒”联盟(T2T)建出了第一个完整的人类参考基因组(被命名为T2T-CHM13)。
相关阅读:《科学》封面重磅:迄今最完整的人类基因组测序结果公布
不过,上述这些都是单个参考基因组的不断升级。原有参考基因组序列的一个重要局限在于,它是由大约20个志愿者的遗传数据拼凑组成的一套基因组,大部分参考序列(约70%)实际上只来自一个志愿者。
“无论你对一个基因组的表征有多准确,都不能代表所有人类群体”,从一开始就参与人类基因组计划、此次也是人类泛基因组参考联盟一员的David Haussler教授解释说,“现在则是一个转折:不再是一个标准人类基因组的基因组学,而是可用于所有人的基因组学。”
此次发布的人类泛基因组参考纳入了T2T的完整基因组序列,还拓展了多样性上的新维度,将来自47个志愿者的全基因组序列集合排列,能方便地分析基因组在个体之间的变化,反映人类物种内部的遗传多样性。
为什么需要更多样化的参考基因组
每个人都是独一无二的,每个人的基因组也略有不同。平均来说,你与另一个人的基因组有大约0.4%的差异。读懂这些微小的差异,可以更好地掌握一个人的健康与疾病状况,有助于疾病的诊断、治疗方法的选择、治疗结果的预测。
当科研人员或临床医生想要根据一个人的基因组找出与疾病相关的变异,他们需要将这个人的DNA与一个作为标准的参照物进行比较,从而确定哪些位置的碱基出现了特殊变化。参考基因组就是这样一个标准,但目前为止的人类参考基因组中基本上每条染色体只有一个序列,这个序列基本上来自一个个体。然而,人类群体中存在大量遗传变异,有些变异在某些特定人群(例如特定祖源)存在,在另一些人群中不存在。
因此,在临床使用基因组学信息——例如预测一种遗传病时,如果想要减少偏差,作为标准的参照物就不能只是某个单一人群的代表,而需要扩大其代表性。
新的泛基因组参考便提供了更多样化、也更准确的标准。研究人员通过复杂的算法,将组装完成的单个基因组序列集合编译为图形结构。如果过去的参考基因组只是一根线,现在的人类泛基因组参考是多个基因组序列的多线并行图。在碱基完全相同的序列,它还是单线形式;而存在人群差异的序列部分,线条则从一根“分化”出地铁轨道般交错的多根,提供了更广泛的参考选择。
人类泛基因组参考联盟的主要研究者之一、德国海因里希海涅大学(Heinrich Heine University)的Tobias Marschall教授指出:“人类泛基因组参考是我们已经等待了十年的一个里程碑,这是在测序技术和生物信息学方面取得了诸多创新后才得以达成的。”
更准确地理解变异和疾病的关系
最新的泛基因组参考图谱比当下的参考人类基因组多了1.19亿个新碱基,其中有大约9000万个碱基来自结构变异。结构变异包括序列的倒置、插入、缺失或串联重复,通常涉及50个以上的碱基,由于其复杂性,过去在使用单一参考基因组的情况下,人类基因组中存在的结构变异70%以上难以识别。而现在,基于更全面的泛基因组参考图谱进行基因组分析,结构变异的检测率可以提高104%,研究人员有机会在未来将结构变异与疾病更好地联系起来。
除了结构变异的检测率提高外,检测较小的遗传变异(例如只有一个或数个碱基差异)时,使用泛基因组参考的准确性也有34%左右的提高。
最新的泛基因组参考图谱还有一项重大突破。我们的染色体成对存在,一套遗传自母亲,一套遗传自父亲,而泛基因组参考包括的单倍型信息——来自47个人的94套基因组序列,可以在分析一个人的基因组时更准确地区分出来自父母的不同染色体。这也将帮助我们更好地理解各种基因和疾病的遗传方式。
圣路易斯华盛顿大学的王艇教授也是该项目的主要研究者之一,他介绍道:"新的泛基因组参考能更准确地发现和评估人类遗传变异,特别是结构变异。人类遗传学和基因组医学的几乎所有领域都可以因此受益。例如,鉴定与人类疾病相关的遗传变异将更加敏感和具体,从而直接改善疾病诊断和治疗。新的参考基因组还为研究遗传变异的功能后果奠定了基础。"
更多新发现
在《自然》专题同时发表的两篇相关论文中,其中一篇聚焦于人类基因组中的单核苷酸变异(SNV)。

美国华盛顿大学医学院的Evan Eichler教授和同事开发了一个SNV图谱,系统评估了片段重复序列(在基因组一个以上位点重复出现、共享高度相同序列的DNA区域)中的SNV,描述了数百万此前未绘制的SNV,并对重复序列和单一序列中出现的SNV进行了比较。

另一篇论文中,美国田纳西大学的研究人员观察了异源着丝粒染色体短臂(中节位于靠近染色体一端)间的重组模式。有关这类染色体之间的DNA交换机制,50年前便已有人提出过假说,却始终因缺乏合适数据而未经观测到。此次,借助泛基因组参考的多对多比较,研究人员终于获得了证据。
下一个里程碑
人类泛基因组参考联盟的研究人员介绍,该项目还在继续采样,目标是纳入350名个体的基因组信息,尤其是目前还未纳入的人群代表,以便扩大多样性和增强不同人群之间的平衡性。他们计划在2024年发布人类泛基因组参考的最终版本,力求代表人类这个物种尽可能多的DNA序列。

王艇教授指出,这不是一个项目的结束,而是一个新领域的开始,以便更有意义地将人类多样性纳入生物学、生物医学和临床科学。新的人类参考基因组将继续增长、扩大和打磨,从而更准确地描绘我们这个物种的生命蓝图——这需要全世界的努力。
人类参考基因组问世二十多年,尽管并不完整,却已经在生物医学领域产生了深远的影响;随着包容性更强、多样性更全面的泛基因组参考上线,下一个十年、二十年,这一成果必将通过临床研究、药物开发和医疗实践的发展造福全球。
参考资料:
[1] Wen-Wei Liao et al., (2023) A draft human pangenome reference. Nature Doi: https://doi.org/10.1038/s41586-023-05896-x
[2] Mitchell R. Vollger et al., (2023) Increased mutation and gene conversion within human segmental duplications. Nature Doi: https://doi.org/10.1038/s41586-023-05895-y
[3] Andrea Guarracino et al., (2023) Recombination between heterologous human acrocentric chromosomes. Nature Doi: https://doi.org/10.1038/s41586-023-05976-y
[4] Glenn Hickey et al., (2023) Pangenome graph construction from genome alignments with Minigraph-Cactus. Nature Biotechnology Doi: https://doi.org/10.1038/s41587-023-01793-w
[5] Arya Massarat et al., (2023) A collective human reference genome. Nature
[6] Human pangenome reference will enable more complete and equitable understanding of genomic diversity. Retrieved May 10, 2023 from https://www.eurekalert.org/news-releases/988471
本文来自微信公众号:学术经纬 (ID:Global_Academia),作者:药明康德内容团队