
本文来自微信公众号:学术经纬 (ID:Global_Academia),作者:药明康德内容团队,头图来自:视觉中国
2008年,《自然》发布了首个急性髓系白血病(AML)样本的全基因组序列。科学家使用下一代测序技术,对一名AML患者的肿瘤细胞和正常皮肤细胞样本进行了全基因组测序并进行比较,从而找到了癌细胞中的8个全新基因突变。当时的这一突破性研究验证了肿瘤学家的猜测,那就是利用基因组测序,能够发现可能导致肿瘤发生的全新基因突变,从而提供一系列潜在的药物靶点。
十多年来,随着测序技术的突飞猛进,癌症领域的测序研究以惊人的速度进展,为人们理解癌症的发生和开发抗癌疗法提供宝贵信息。
今日,顶尖学术期刊《科学》的最新一期上,迄今为止样本规模最大的一项全基因组测序研究,分析了12000多名癌症患者的全基因组序列,揭示出数十种过去未知的肿瘤突变特征,进一步扩宽人们对癌症发病原因的认识。
研究人员还为每种癌症类型创建了常见突变特征与罕见突变特征的概念,为未来的个体化癌症治疗提供重要指导。

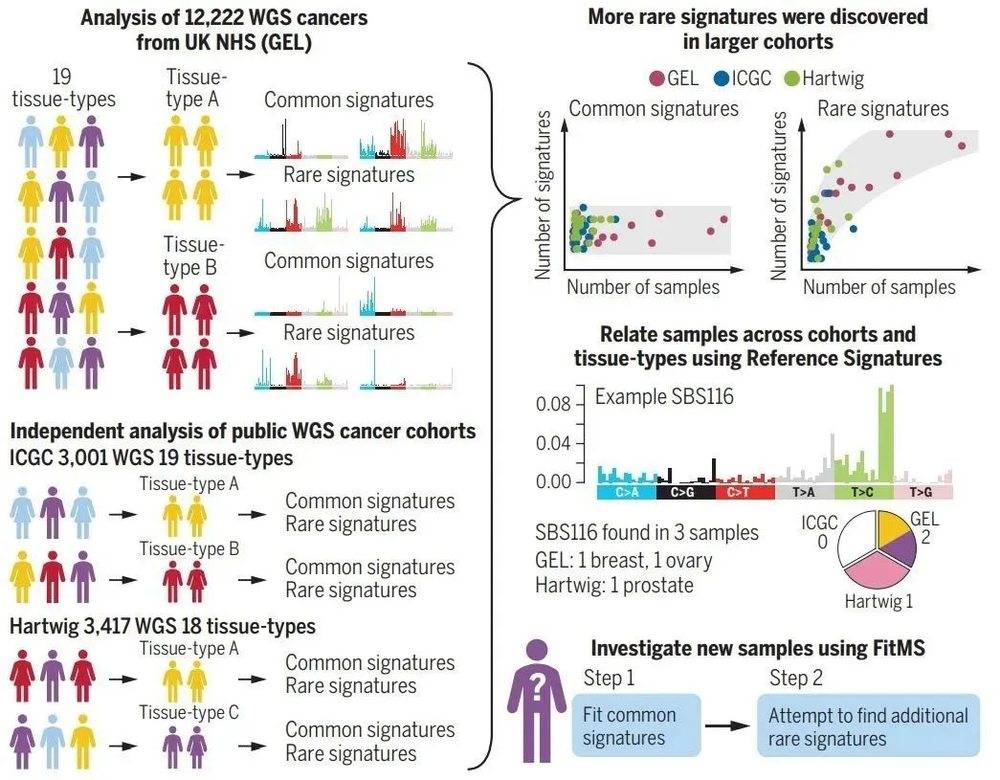
为这项研究提供数据基础的是英国的“十万基因组计划”(100000 Genomes Project)。该计划针对英国国家医疗服务体系(NHS)登记在册的约8.5万名癌症或罕见病患者进行全基因组测序。
在此次发表的研究工作中,科学家们分析了其中12000多名癌症患者的全基因组序列,检测每个癌症患者的DNA上所有可能导致癌变的变化,进行突变特征(mutational signatures)的分析。
突变特征指的是每个癌症患者经历的DNA损伤与修复过程的印记。这些信息可以透露患者过去是否接触过导致癌症的环境因素(例如吸烟或紫外线辐射等)或是细胞内部发生了什么障碍。打个比方来说,突变特征就像罪犯在犯罪现场留下的指纹,有助于查明癌症发生的罪魁祸首。
每种癌症都有数千个突变。研究人员在人体的乳腺、肺部、神经系统等十几个组织器官中分别鉴定了不同的突变特征,寻找不同癌症类型的患者具有的共性和差异,将常见的突变过程与人群中发生频率较低的罕见突变过程区分开来。然后,研究团队使用了包含数千名癌症患者的两个队列数据对他们的发现进行验证。
由此,研究团队不仅证实了许多先前报道过的肿瘤突变特征,还识别出58个过去未知的肿瘤突变特征,包括40个新的单碱基置换(SBS)和18个双碱基置换。这些新发现的突变特征为我们理解癌症为什么会发生提供了全新的线索。
![▲常见突变特征和罕见突变特征的发现和应用(图片来源:参考资料[1])<br label=图片备注 class=text-img-note>](https://i.aiapi.me/h/2022/04/22/Apr_22_2022_11_20_14_37624984923583698.jpeg)
根据这些分析结果,研究人员还确认了每个器官都包含数量有限的常见突变特征(通常是5~10个单碱基置换)。与此同时,每种类型的肿瘤可能还具有一些罕见突变特征。
基于这些数据,研究人员创建了一种名为FitMS的新算法,其他研究者或是临床医生每次获得新的肿瘤样本后,可以用来识别已知的常见突变特征,以及找出额外的罕见突变特征。
展望未来,研究人员希望这种方法最终将有助于每一名癌症患者在临床治疗中获得更好的诊断和治疗方案,比如是否会对免疫疗法有反应、受益于哪种靶向治疗等。
参考资料:
[1] Andrea Degasperi et al., (2022) Substitution mutational signatures in whole-genome–sequenced cancers in the UK population. Science. Doi: https://doi.org/10.1126/science.abl9283
[2] Largest study of whole genome sequencing data reveals ‘treasure trove’ of clues about causes of cancer. Retrieved Apr. 21, 2022 from https://www.eurekalert.org/news-releases/949983
本文来自微信公众号:学术经纬 (ID:Global_Academia),作者:药明康德内容团队