
本文来自微信公众号:原理(ID:principia1687),作者:小雨,头图来自:DeepMind
一
在21世纪面临的重大挑战中,无论是研究如何生产清洁电力,还是开发高温超导体,都需要我们能够设计出特定性能的新材料。如果要想在计算机上完成这项工作,就需要对电子进行模拟,因为这些亚原子粒子主导了原子如何结合成分子,同时也负责着固体中的电流流动。
可以说,了解分子内的电子的位置,对于解释分子的结构、性质和反应性等特征都至关重要。尽管科学家已经为此付出了数十年的努力,也曾取得过一些重大的进展,但至今为止,想要准确地模拟电子的量子力学行为,仍然是一个艰巨的挑战。
现在,在一篇新发表于《科学》杂志上的论文中,DeepMind研究团队介绍了一个新的神经网络——DM21,能够通过预测分子内电子的分布来了解分子的特征。它可以比现有技术更准确地计算一些分子的性质。
二
理论上说,材料和分子的结构完全由量子力学决定。在近一个世纪以前,物理学家埃尔温·薛定谔 (Erwin Schrödinger)提出了著名的薛定谔方程,描述了量子力学粒子的行为。但是,想要将这个方程应用于分子中的电子,是非常困难的。因为所有的电子都会相互作用,这就使得基于薛定谔方程来计算分子结构或分子轨道变得异常困难,它似乎需要我们能够追踪每个电子的位置。这样一项工作,即使是对于只有少量电子的情况,也是一项噩梦般复杂的任务。
一次重大的进展出现在上世纪60年代。当时,理论物理学家皮埃尔·奥昂贝格(Pierre Hohenberg)和沃尔特·科恩(Walter Kohn)意识到,并没有必要对每个电子的行为都进行单独追踪,只要知道任意电子在每个位置的概率,即电子密度,就足以精确计算所有的相互作用了。
在证明了这一点后,科恩发展出了密度泛函理论(density functional theory,DFT),它能够帮助我们对分子中的电子分布进行精确近似。虽然DFT在本质上涉及到一定程度的近似,但它是唯一一种可用于研究物质在微观水平上如何以及为何以某种方式表现的实用方法。因此自60年代开始,DFT便成了所有科学领域中应用最广泛的技术之一。科恩也因此获得了1998年的诺贝尔化学奖。
然而,这种技术有其明显的局限性。虽然它证明了电子密度和相互作用能之间存在映射关系,但50多年来,这种映射的确切性质仍是未知的,必须通过近似。数十年来,不断有研究人员为密度泛函提出不同精度的近似,但这些近似方法都存在各自的系统性误差,有时会导致DFT失效。比如它们会对某些类型的分子给出错误的结果,在一个著名的例子中,DFT方法就错误地预测了氯化钠,使得一个氯原子即使与钠原子相距无限远,氯原子也仍保留着钠原子的一个电子的部分。
三
现在,DeepMind的研究人员通过将这个泛函表示为一个神经网络,成功地实现在无系统错误的情况下学习这些泛函。
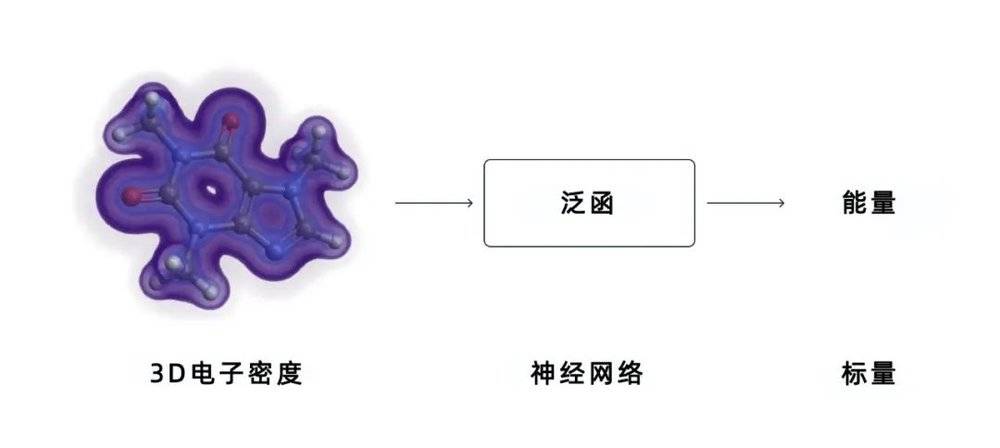
在研究过程中,他们用从薛定谔方程中得到的1161个精确解的数据,对神经网络进行了训练。为了提高准确性,他们还将一些已知的物理定律整合到网络中。然后,他们用一组分子来测试训练过的系统,得出了令人惊叹的结果。新的研究结果表明,这种新的人工智能模型能够比传统的DFT方法做出更准确的预测,更好地描述一系列化学反应。
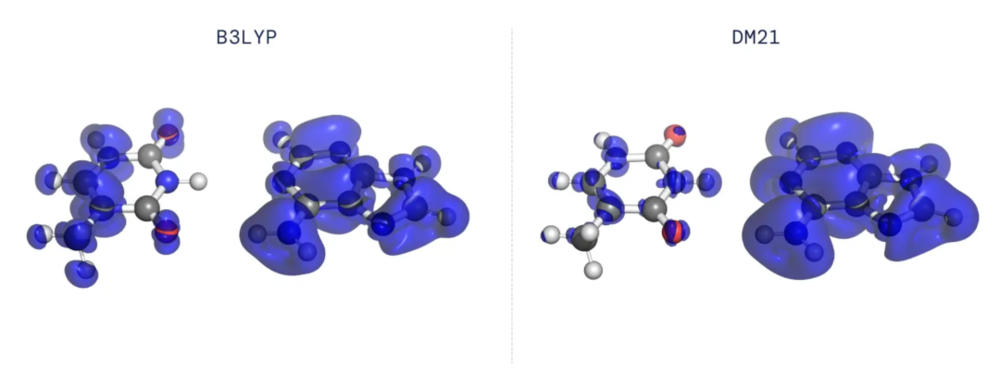
尤为值得一提的是,在这次的研究结果中,研究人员解决了长期存在与传统泛函中的两个问题。
第一个问题名为退局域误差:在DFT计算中,泛函会通过找到能够使能量最小化的电子构型,来决定分子的电荷密度。因此,泛函中的误差会造成计算出的电子密度中出现误差。大多数现有的对密度泛函的近似,都不会让电子密度精准地局限在一个分子或一个原子周围,而是倾向于让电子密度以不切实际的形式分散在几个原子或几个分子之周围。
另一个问题被称为自旋对称性破缺:当描述化学键的破坏时,现有的泛函趋向于不现实地偏向一种基本对称性被打破的构型,这种基本对称就是自旋对称性。由于对称性对于我们理解物理和化学起着至关重要的作用,因此这种人为的破坏对称性,成了现有泛函的一个主要问题。
从理论上看,任何涉及电荷运动的化学-物理过程,都容易发生退局域误差;而任何涉及到键的断裂的过程,都容易发生自旋对称性破缺。电荷的运动和键的断裂是许多重要技术应用的核心,但这也可能导致泛函在描述一些最简单的分子时出现严重问题,正如我们在前文所提到的氯化钠的例子那样。
DFT作为如此重要的一项技术,因此不难理解科学家会在期盼这些泛函能够解释更复杂的分子相互作用之前,必须先做到能确保简单化学的正确性。通过使用神经网络来表示泛函,并训练数据集来捕获期望的精确泛函的分数电子行为,新的研究解决了这两个问题。他们的泛函在广泛的、大规模的基准测试中,都显示出了高度的准确性。
四
计算机模拟在现代工程中发挥着核心作用,使人们有可能为各种实际的应用问题提供答案。
我们已经用它们来模拟桥梁是否能支撑、火箭能否能升空等问题,并随着技术越来越多地转向量子尺度时,希望它们也能帮我们探索关于材料、药物和催化剂的问题,以及很多我们从未见过甚至难以想象的问题。
现在,深度学习已经显示出了在量子力学水平上精确模拟物质的能力。让人工智能来计算电子密度,或许是DeepMind团队迄今为止进行过的最雄心勃勃的尝试,这是DFT计算的终极结果。
参考来源:
https://deepmind.com/blog/article/Simulating-matter-on-the-quantum-scale-with-AI
https://www.nature.com/articles/d41586-021-03697-8
https://cen.acs.org/physical-chemistry/computational-chemistry/Machine-learning-solves-long-standing/99/web/2021/12
本文来自微信公众号:原理(ID:principia1687),作者:小雨