
目前市售的传统抗抑郁药物通常需要几周甚至几个月后才能起效,并且对三分之一的难治性抑郁症患者无改善作用。近年来,多种致幻剂在抑郁症治疗方面展现了潜能。从“神奇蘑菇”中提取的天然致幻剂裸盖菇素(Psilocybin),于2019年被美国FDA授予作为重度抑郁症和药物抵抗性抑郁症的突破性疗法。
二期临床研究结果表明,裸盖菇素单次用药一天内即可极大改善抑郁症患者症状,且效果可持续三个月以上。但是,致幻剂的致幻副作用限制了其临床研究和应用。因此,科学家长期致力于寻找和研发非致幻且能快速起效的新型抗抑郁药。
1月28日,中国科学院分子细胞科学卓越创新中心(生物化学与细胞生物学研究所)汪胜研究组与上海科技大学iHuman研究所程建军研究组合作在Science上,发表了题为Structure-based discovery of non-hallucinogenic psychedelic analogs的研究论文。该工作从解析致幻剂与其靶点血清素2A受体(5-HT2AR)的复合物结构出发,发现致幻剂除了先前预测的与5-HT2AR的经典结合模式之外,存在另一种位于经典结合模式上方的、受脂质调控的结合模式。研究人员发现在脂质分子存在下,致幻剂的第二种结合模式对于5-HT2AR下游β-arrestin信号的偏好性激活十分重要;进一步实验结果表明,致幻剂的致幻效果与G蛋白和β-arrestin两条下游信号同时高效率激活密切相关。基于第二种结合模式,研究人员设计并合成了以IHCH-7086和IHCH-7079为代表的一系列5-HT2AR的β-arrestin信号偏好性激动剂;在动物体内实验中证明该系列化合物无致幻作用,但具备与致幻剂相似的快速抗抑郁效果。
Psilocybin(裸盖菇素)在人体内被代谢为psilocin(脱磷酸裸盖菇素),后者与5-HT2AR结合发挥作用。汪胜研究组首先解析了5-HT2AR与内源性配体serotonin(血清素)、致幻剂psilocin、致幻剂LSD(麦角酸二乙基酰胺),以及非致幻剂lisuride(麦角乙脲,帕金森病药物)等四种复合物的晶体结构。LSD和lisuride的麦角灵骨架的结合位置与实验室前期解析的其他麦角灵药物类似,均结合在正位结合位点(orthosteric binding pocket,OBP);与之前预测的正位结合位点不同,serotonin和psilocin受脂质调控结合在OBP上方的延伸性口袋(Extended binding pocket,EBP)。进一步实验结果表明该脂质为monoolein(甘油单油酸酯),可占据5-HT2AR独有的侧边口袋(side-extended pocket,SEP),并与TM5的残基有一定极性相互作用。实验表明monoolein通过G2385.42残基位置进入OBP,激活5-HT2AR下游的G蛋白信号并且该信号的激活可以被5-HT2AR的选择性抑制剂MDL100907所抑制。
研究人员认为serotonin和psilocin在5-HT2AR中的两种不同结合形式(OBP和EBP),可能对受体功能产生不同影响。实验发现单独monoolein无法激活5-HT2AR下游的β-arrestin信号,但在serotonin存在的情况下可剂量依赖性地激活β-arrestin信号。相比之下,LSD存在的情况下,未能深入OBP的monoolein则不能激活β-arrestin信号。结果表明serotonin和psilocin的第二种结合模式对其β-arrestin信号的激活十分重要。
5-HT2AR-LSD和5-HT2AR-lisuride的结构比对表明,LSD中两个乙基均接触EBP中的残基Y3707.43,而lisuride只有一个乙基与其相互作用。实验发现5-HT2AR-Y3707.43W突变体能显著提高lisuride的β-arrestin信号,而对于G蛋白信号影响不大。然而由于Y3707.43直接与LSD的两个乙基相互作用,Y3707.43W突变体同时降低了LSD介导的G蛋白信号和β-arrestin信号。结果表明虽然LSD和lisuride在OBP中结合模式相似,但在EBP中结合模式的不同导致其与TM7的相互作用不同,从而影响了下游的β-arrestin信号。
早期研究表明,致幻剂对5-HT2AR的亲和力与其精神活性密切相关。动物行为模型不能精确捕捉到由致幻剂在人类中产生的知觉、认知和情绪的扰动。然而,研究表明,小鼠头部抽搐反应(Head twitch response,简称HTR)与人类产生致幻剂诱导产生的幻觉密切相关。研究人员进一步通过搭建的基于磁信号检测小鼠HTR的模型中验证了5-HT2AR-Y3707.43W点突变小鼠可以成功抹去LSD的HTR效应,同时保留了LSD的抗抑郁效果。结果表明致幻剂的精神活性作用需要5-HT2AR介导高效率的信号转导,且致幻剂的致幻效果和抗抑郁效果可通过改变5-HT2AR激动剂的药理学活性而分离。
通过上述数据,研究人员推测靶向EBP可以增强5-HT2AR介导的β-arrestin信号,有利于设计出β-arrestin信号偏好性配体。实验室前期的研究结果表明抗精神病药物的头部基团均与其GPCR靶向受体的EBP相结合,并且5-HT2AR是非典型抗精神病药物的重要靶点之一。通过分析市售的几十种抗精神病药物,研究人员锁定了2019年FDA批准上市的lumateperone(卢美哌隆,5-HT2AR的抑制剂),并解析了其与5-HT2AR的复合物晶体结构,发现lumateperone头部基团的四环骨架结合在5-HT2AR的EBP。研究人员基于结构设计并合成了头部基团IHCH-7113。进一步数据表明,IHCH-7113具有微弱的β-arrestin信号偏好性且在5-HT2AR两条下游信号通路中均具有较高的激活效率,可诱导小鼠产生HTR。
研究人员进一步通过基于磁信号检测小鼠HTR的模型中检测了IHCH-7086,IHCH-7079(IHCH-7086类似物)的致幻效果,结果表明具有明显的β-arrestin信号偏好性的IHCH-7086和IHCH-7079在注射高剂量(10 mg/kg)的情况下不会诱导小鼠产生HTR。随后,研究人员在不同方法构建的小鼠抑郁样模型中验证了IHCH-7086和IHCH-7079的抗抑郁效果,实验结果表明IHCH-7079和IHCH-7086与先前已报道的LSD所具有的抗抑郁效果类似,能够明显改善小鼠的抑郁样行为,并且5-HT2AR的选择性抑制剂MDL100907能抑制其改善作用。
进一步通过以结构为导向的化合物理性设计,研究人员在IHCH-7113的基础上引入甲氧基苯基合成了IHCH-7086及其系列衍生物;使其相对lumateperone的氟苯基避开5-HT2AR的PIF结构域(决定了lumateperone的抑制剂效果);按照设计,该系列化合物将以结合EBP为主(β-arrestin偏好),同时避开结合OBP的脂质结合口袋(G 蛋白偏好)。通过解析IHCH-7086和5-HT2AR的复合物晶体结构,IHCH-7086在5-HT2AR中的结合模式的确如研究人员所设想,且IHCH-7086表现出明显的β-arrestin信号偏好性。
该研究深入阐明了致幻的分子机制,有助于理解致幻剂缓解抑郁症的药理机制,为新型速效、长效抗抑郁药物的研发提供了理论指导。研究工作得到科技部重点研发项目、中科院战略性先导科技专项、国家自然科学基金、科技部青年千人基金、上海市科学技术委员会启明星计划项目等的资助。研究工作得到分子细胞卓越中心化学生物学技术平台和动物实验技术平台的支持。
论文链接
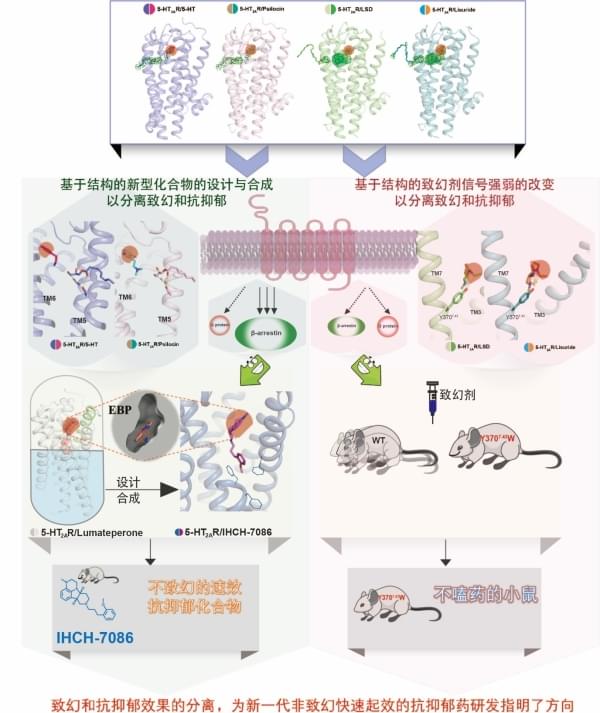
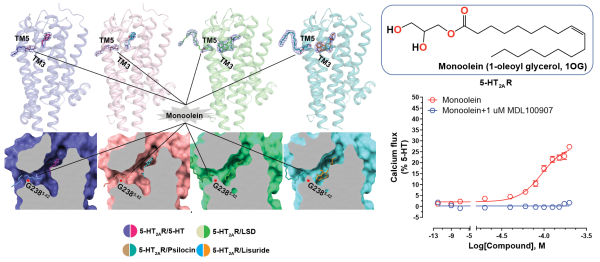
图1.脂质对5-HT2AR的激活
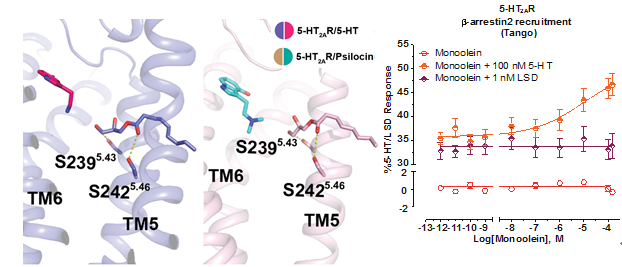
图2.脂质存在下serotonin和psilocin结合在EBP中
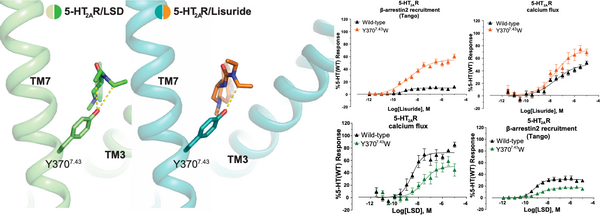
图3.Y3707.43对lisuride和LSD激活5-HT2AR下游信号的影响
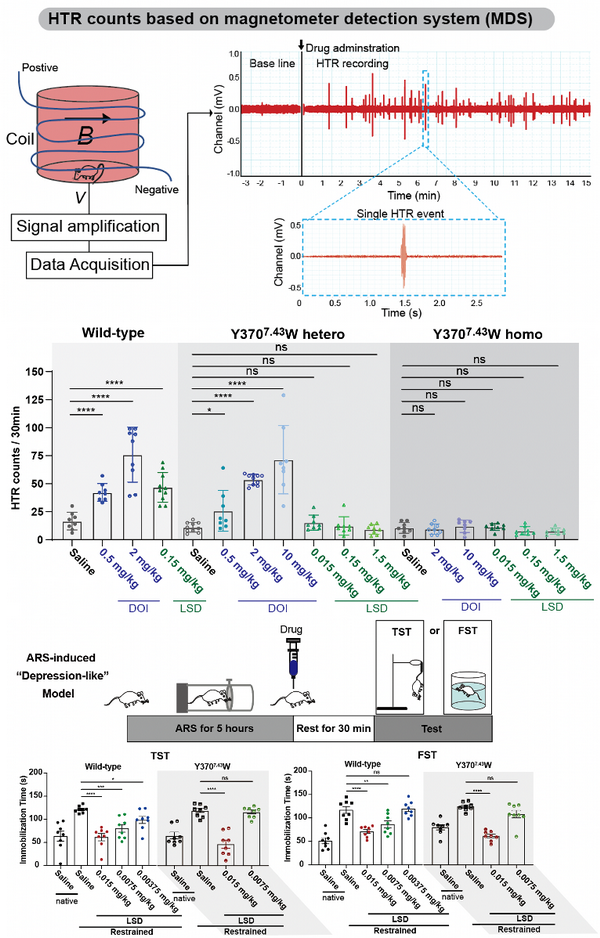
图4.5-HT2AR-Y3707.43W小鼠对LSD的致幻和抗抑郁效果的影响
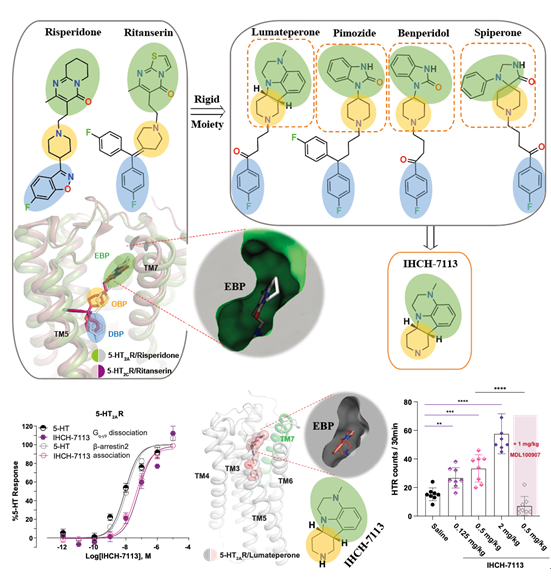
图5.IHCH-7113的结构设计思路,激活信号和致幻效果
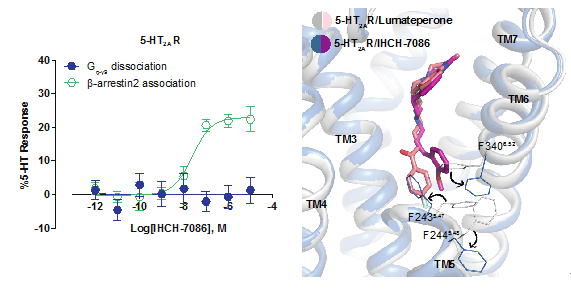
图6.IHCH-7086的激活信号和与5-HT2AR结合的复合物结构
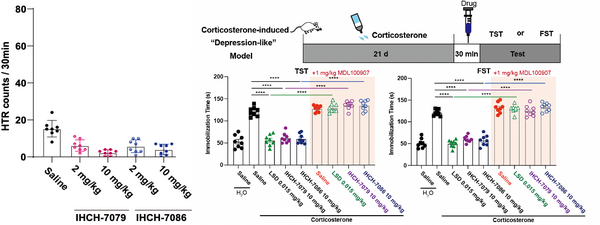
图7.IHCH-7086和IHCH-7079的致幻和抗抑郁效果